L’attività di ricerca del gruppo è focalizzata sulle basi molecolari del cancro. Puntiamo a contribuire alla comprensione dei meccanismi molecolari che determinano l’aggressività del cancro, così come i meccanismi che regolano la complessa interazione tra cellule tumorali e microambiente, incluse altre cellule non tumorali, ed il sistema immunitario. Il nostro approccio è quello di studiare le interazioni funzionali tra geni oncosoppressori e vie di segnalazione che controllano attività fisiologiche e patologiche delle cellule.
1) Comprendere le proprietà oncogeniche delle mutazioni “gain-of-function” di p53 attraverso lo studio di nuove interazioni proteiche
Il gene oncosppressore TP53 è mutato molto spesso nei tumori umani. Circa il 70% delle mutazioni sono missenso e causano la sostituzione di un singolo aminoacido. Quindi, molti tumori esprimono forme aberranti della proteina p53 (mutp53) che non solo perdono la normale funzione oncosoppressiva, ma acquisiscono nuove proprietà oncogeniche (un fenomeno definito “gain of function”), in quanto promuovono la progressione tumorale e la chemioresistenza. In accordo, tumori con p53 mutata tendono ad essere più aggressivi e spesso sono associati a prognosi peggiore.
Il meccanismo della “gain-of-function” in parte dipende dall’interazione di mutp53 con altre proteine. E’ infatti plausibile che le mutazioni oncogeniche possano alterare drasticamente il profilo di interazione della p53; per esempio, perdendo il legame con certe proteine ma preservando l’interazione con altre. Oppure, p53 mutata può formare nuovi complessi con proteine cellulari che normalmente non interagiscono con la p53 wild-type. Per questo motivo, c’è un considerevole interesse a definire il profilo delle interazioni proteiche dei mutanti oncogenici di p53.
Alcuni anni fa abbiamo condotto uno screening in vitro per scoprire proteine che interagiscono con la singola p53 (Dmp53) codificata nel genoma di Drosophila melanogaster. Partendo dai nuovi interattori di Dmp53, abbiamo analizzato le corrispondenti proteine umane, saggiandone la possibile interazione con tutti i membri della famiglia: p53, p63 e p73. Abbiamo così identificato 37 potenziali nuovi partner funzionali di questa importante famiglia di oncosoppressori (Lunardi et al, 2010). Abbiamo poi saggiato i nuovi interattori per verificare la loro possibile associazione con p53 mutata (H175R), scoprendo che la maggior parte di essi lega efficientemente mutp53 (dati non pubblicati). Attualmente, stiamo esplorando il possibile ruolo funzionale di queste interazioni nel quadro della “gain of function” dei mutanti oncogenici di p53.
Recentemente abbiamo scoperto che l’espressione di forme mutate di p53 conferisce maggiore resistenza allo stress del reticolo endoplasmico (ER) modificando la risposta trascrizionale UPR (Unfolded Protein Response) nelle cellule tumorali. Abbiamo osservato che mutp53 inibisce l’attivazione degli effettori pro-apoptotici dell’UPR, mentre promuove l’attivazione del fattore ATF6, che favorisce la sopravvivenza in condizioni di stress del ER. I dati suggeriscono che l’inibizione farmacologica di mutp53 può essere combinata con l’inibizione di ATF6 per aumentare la sensibilità delle cellule tumorali alla chemioterapia (Sicari et al., 2019).
2) L’oncosoppressore DAB2IP come bersaglio per inattivazione post-trascrizionale nel cancro
L’interazione dinamica e lo scambio di informazioni tra cellule tumorali e cellule della matrice stromale influenzano in modo determinante l’aggressività del cancro. In questo contesto, proteine di segnalazione che controllano specificità, ampiezza, e durata delle risposte cellulari agli stimoli provenienti dall’esterno possono svolgere un ruolo fondamentale, che è spesso sottovalutato.
Appartiene a questa categoria di “modulatori” l’oncosoppressore DAB2IP/AIP1. Si tratta di una proteina citoplasmatica che agisce da inibitore di Ras (Ras-GAP), ma controlla anche l’attivazione di NF-kB da parte di citochine infiammatorie, e l’attivazione di PI3K-AKT da parte di vari fattori di crescita. Ci sono evidenze convincenti che la perdita di funzione di DAB2IP aumenta l’aggressività delle cellule tumorali; infatti, sono stati osservati diversi meccanismi di inattivazione di DAB2IP nel cancro (Bellazzo et al., 2016).
3) Inattivazione di DAB2IP da parte di p53 mutata
Abbiamo scoperto che mutp53 può legare ed inibire DAB2IP nel citoplasma, potenzialmente modificando la risposta cellulare a una varietà di segnali provenienti dall’esterno. Degno di nota, diverse forme mutate di p53 possono legare DAB2IP, indipendentemente dalla specifica mutazione missenso; quindi, la formazione di questo complesso potrebbe essere una caratteristica generale delle p53 mutate, e questo asse molecolare potrebbe diventare un promettente bersaglio per lo sviluppo di terapie mirate.
Dal punto di vista funzionale, abbiamo osservato che mutp53, legando e inibendo DAB2IP, altera la risposta delle cellule tumorali al TNF, rendendole più aggressive in un contesto infiammatorio (Di Minin et al., 2014). In modo analogo, abbiamo scoperto che l’interazione mutp53-DAB2IP altera la risposta delle cellule tumorali all’insulina, stimolando l’attivazione prolungata della chinasi AKT, ed aumentando proliferazione e invasività. Quest’ultima osservazione ha rivelato il potenziale impatto della mutazione di p53 nei tumori al seno in pazienti iperinsulinemiche obese o diabetiche (Valentino et al., 2017).
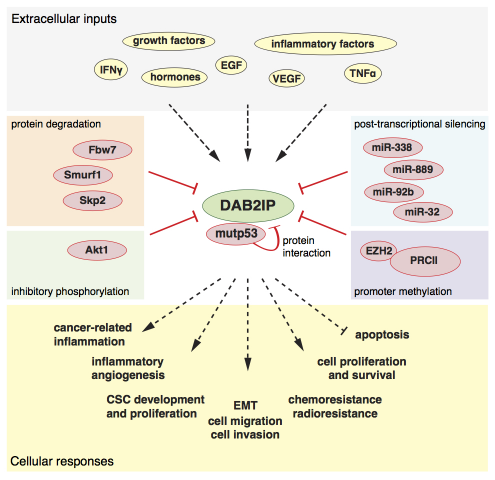
4) Inattivazione di DAB2IP da microRNA
Abbiamo identificato nuovi microRNA in grado di inibire l’espressione di DAB2IP. Tra questi abbiamo caratterizzato il miRNA-149-3p come un potente antagonista dell’espressione e delle funzioni di DAB2IP. L’espressione di miR-149-3p in cellule di tumore prostatico blocca l’attività di DAB2IP, innescando un programma genico pro-angiogenico, ed un comportamento cellulare invasivo. Esperimenti in vivo hanno confermato queste osservazioni; infatti, l’inibizione del miR-149-3p riduce la crescita e la disseminazione di cellule di tumore prostatico in topi nudi e larve di zebrafish (Bellazzo et al., 2018).
Oltre ad avere effetti a livello delle singole cellule trasformate, l’espressione di miR-149-3p può anche agire sul microambiente tumorale. Infatti, abbiamo scoperto che miR-149-3p viene secreto dalle cellule tumorali e induce la riduzione dei livelli DAB2IP in cellule riceventi non trasformate, in particolare in cellule vascolari endoteliali. Tramite questa regolazione inter-cellulare, le cellule cancerose possono teoricamente inattivare DAB2IP nelle cellule dello stroma, generando così un microambiente favorevole alla crescita e disseminazione del tumore. Sappiamo che il miR-149-3p contribuisce a questa azione in line tumorali di prostata, ma è molto probabile che altri fattori siano coinvolti in questo fenotipo - inclusi altri miRNA.
Al momento siamo interessati a comprendere i meccanismi ed il potenziale impatto dell’inibizione inter-cellulare di DAB2IP in cellule endoteliali, ed eventualmente in altre cellule stromali. Infatti questa conoscenza potrebbe ampliare la nostra visione del complesso rapporto di regolazione.





