Research
We are interested in the molecular basis of cancer. We aim to help understand the molecular mechanisms that drive cancer progression and aggressiveness, as well as the mechanisms that regulate the complex interaction of cancer cells with their environment, including other cells and the immune system. Our approach is to look at functional interactions between tumor suppressor genes and signaling pathways that control physiological and pathological cell activities.
1) Exploring the gain-of-function properties of mutant p53 through novel interacting proteins
In human cancers, tumor suppressor gene TP53 is mutated very frequently. Notably, about 70% of TP53 mutations are missense, inducing single aminoacid substitutions in the p53 protein. Therefore, many tumors express mutant p53 proteins that not only lose their normal tumor-suppressive functions, but also acquire novel oncogenic properties (a phenomenon called gain-of-function), as they in fact promote cancer progression and drug resistance. Accordingly, tumors with mutant p53 tend to be more aggressive, and are often associated with worst prognosis.
The mechanism underlying the gain of function partly depends on the interaction with other cellular proteins. In fact, tumor-associated p53 mutations may drastically alter the protein interaction profile of p53. For instance, they may prevent interaction with certain proteins, while binding to other proteins may remain unaffected. Alternatively, mutant p53 (mutp53) could acquire the capability to interact with cellular proteins that normally do not bind wild-type p53. For this reason, there is considerable interest in defining the interaction profile of oncogenic p53 mutants as compared to that of wild-type p53.
Various years ago, we searched the Drosophila genome for proteins that interact in vitro with the single p53 protein (Dmp53) in this organism. We reasoned that an unbiased "phylogenetic" approach could reveal novel, highly conserved molecular circuits regulating this very important tumor suppressor pathway in mammals. We used the Drosophila hits as a framework to study the corresponding interactions in mammals. We tested the human orthologs of 41 newly discovered Dmp53 interactors for association with human p53, p73 and p63, and obtained a list of 37 potential partners of these crucial tumor suppressor proteins (Lunardi et al., 2010).
As a follow-up of this work, we systematically tested the novel interactors for their binding to mutant p53(H175R). Notably, a consistent fraction of the proteins bound to mutant p53 (unpublished data). Thus, we are studying the possible role of these mutp53 interactors in the gain of function phenotype associated to oncogenic p53 mutants.
Recently, we discovered that expression of mutant p53 enhances the resistance to ER stress in cancer cell by modulating Unfolded Protein Response (UPR). We found that mutp53 inhibits activation of pro-apoptotic branches of the UPR, while it enhances activation of the pro-survival UPR effector ATF6. Data suggest that pharmacological inhibition of mutp53 could be combined with ATF6 inhibitors to sensitize cancer cells to chemotherapy (Sicari et al., 2019).
2) The tumor suppressor DAB2IP as a target for post transcriptional inactivation in cancer
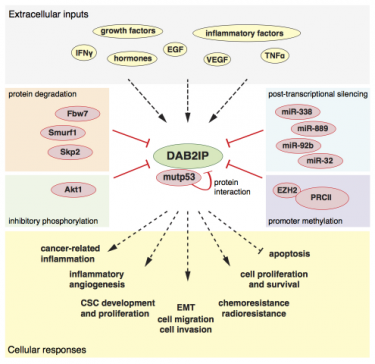
The dynamic crosstalk between tumor cells and tumor stroma is a major determinant of cancer aggressiveness. In this context, signaling modulators that control specificity, amplitude, and duration of cell responses to extrinsic inputs can play a crucial role - often underestimated.
The tumor suppressor DAB2IP/AIP1 belongs to this category; is a cytoplasmic Ras inhibitor (Ras-GAP) that also restrains NF-kB activation by inflammatory cytokines, and PI3K-AKT activation by various growth factors. There are solid evidences that DAB2IP loss-of-function in cancer cells enhances their aggressiveness; accordingly, various mechanisms for DAB2IP inactivation have been described in human cancers (Bellazzo et al., 2016).
3) DAB2IP inactivation by mutant p53
We discovered that mutp53 can bind and inhibit the tumor suppressor DAB2IP, thus potentially affecting the cell’s response to a variety of extracellular inputs. Interestingly, it appears that various different tumor-associated p53 mutants can bind DAB2IP, regardless of the specific missense mutation; this suggests that this interaction may be a general feature of mutp53 proteins, so this molecular axis may be an attractive target for therapeutic strategies.
Functionally speaking, we found that mutant p53, by binding DAB2IP in the cytoplasm, alters the response of cancer cells to TNF, increasing their aggressiveness in response to inflammation (Di Minin et al., 2014). We also found that the mutp53-DAB2IP interaction affects the response of cancer cells to Insulin, causing enhanced and prolonged AKT activation. This finding revealed a potential impact of p53 mutation in breast tumors that develop in hyperinsulinemic obese or diabetic patients (Valentino et al., 2017).
4) DAB2IP inactivation by micro-RNA
We have identified a panel of microRNAs able to target the mRNA of DAB2IP. Among them, we characterized miRNA-149-3p as a potent antagonist of DAB2IP translation and functions. miR-149-3p expression in prostate cancer cell lines counteracts DAB2IP action, favoring an immunogenic and pro-angiogenic NF-kB-related gene expression program, and an invasive phenotype. In vivo xenograft experiments supported the in vitroresults: inhibition of miR-149-3p counteracted prostate cancer cell dissemination in nude mice, and reduced cancer cell growth and angiogenic remodeling in zebrafish larvae (Bellazzo et al., 2018).
In addition to inducing cell-autonomous effects, expression of miR-149-3p in cancer cells can possibly act also on the tumor microenvironment. We found that secreted miR-149-3p induces DAB2IP downregulation in non-transformed cell lines and in primary endothelial cells. This is a possible mechanism by which cancer cells could inactivate DAB2IP in stromal cells, to establish a supporting microenvironment that would facilitate tumor growth and dissemination. We found that miR-149-3p partially mediates this phenotype in prostate cancer cell lines, but additional factors are likely to be involved, including other DAB2IP-targeting miRNAs.
We are currently interested in understanding the mechanisms and potential impact of cell non-autonomous DAB2IP inhibition in endothelial cells, and possibly in other stromal cells. This may provide further insight on the reciprocal regulation between cancer and stroma, and may reveal novel targets for therapy.


