Our research focuses on the comparative genomics of the animal immune system. Diversity at immune genes is necessary for long term survival of species, populations and individuals. Our research will largely exploit the analysis of next generation sequence data, including transcriptome assembly and annotation, sequence homology predictions and phylogenetic analyses.
Most of our research focuses on the early natural history of the invertebrate innate immune system (Bivalves and Crustaceans), with particular attention given to the genetics of antimicrobial peptides (AMP), pathogen receptors (PRR) and the relative signal transduction. Our long standing expertise on genomic data management paved the way for long term collaborations with colleagues interested in the comparative genomics of vertebrates, in particular fishes. In recent years, our collaborations have supported the analysis of both animal and plant biodiversity. For this purpose we apply both DNA metabarcoding and environmental DNA (eDNA) technologies.
Genomics of bivalve immunity
(Researchers involved: Alberto Pallavicini, Marco Gerdol)
The evolution of innate defense systems exposes the never-ending race to arms between animal hosts and more quickly evolving pathogenic microorganisms. Coastal waters are subject to relevant changes in temperature, salinity and oxygen availability due to seasonal variations, which can result in significant alterations of the microbial communities, occasionally leading to an increased presence of bacteria and to harmful algal blooms. In addition, costal marine waters are among the most heavily impacted environments by anthropic activities and pollution by heavy metals, fertilizers, hydrocarbons, etc.
Marine filter-feeding bivalve mollusks have developed specific defense strategies to deal with these potential hazards. In particular the mussel Mytilus galloprovincialis, an important seafood in the Mediterranean area, is relatively tolerant to a wide range of pathogens and environmental changes, thus being often selected as a sentinel species in ecotoxicological investigations. However, even though mortality events caused by infective agents and parasites apparently occur less frequently in mussels than in other bivalves, the molecular bases of such tolerance are unknown.
The main aim of this project is to elucidate the molecular players involved in the bivalve immune response, using a combination of classical molecular biology techniques and –omic approaches. This topic is of particular interest in invertebrates, which are organisms unable to mount long-term responses due to the lack of an adaptive immune system. The application of next generation sequencing technologies to the study of mussel transcriptome has already permitted to evidence that massive events of gene family expansion and the fast diversification rate of immune receptors and effectors are essential for pathogen sensing and targeting in this species. The main lines of research in this project concern the in depth study of the variability and the regulation of mussel pattern recognition receptors (PRRs), antimicrobial peptides (AMPs) and of the signal transduction pathways involved in the bivalve innate immune response to bacteria, viruses and other parasites.
We have recently demonstrated, for the first time in the animal kingdom, the presence of an open pan-genomes, characterized by a very high number of dispensable genes subjected to presence-absence variation. Such genes might have a key adaptive role, largely explaining the high resilience of this bivalve towards multiple biotic and abiotic stressors, as well as its invasiveness.
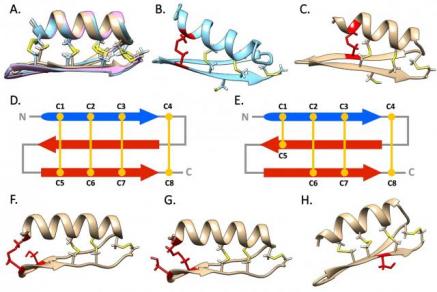
Structural diversity of mytilin-like peptides in marine mussels
Comparative and evolutionary genomics
(Researchers involved: Alberto Pallavicini, Marco Gerdol)
The increasing accessibility of next generation sequencing technologies now permits the analysis of non-model organisms which have been so far almost completely neglected in genetic and genomic studies due to a series of heterogeneous factors including excessive costs, huge genome sizes and difficult maintenance of specimen in laboratory conditions. Over the past few years, our research team has studied species of high interest in the field of evolutionary biology, such as coelacanth and lungfish.

Coelacanth, a “living fossil” though to be extinct until the late ‘30s, and lungfish do indeed cover a key position in the evolutionary tree of tetrapods, and their study can help to clarify several aspects that are still poorly understood about the “water to land” transition. Our activities in this field include the international collaboration in the Latimeria chalumnae genome sequencing consortium and the investigation of the molecular mechanisms at the base of sex determination and transposable element activity.
More recently, our involvement in a national PRIN project in the field of conservation genomics gave us the opportunity to extend our genomic analyses to five endemic Italian species at risk of extinction, which include the iconic Marsican bear and the Adriatic sturgeon. Obtaining the complete high quality reference genome sequences of such species will provide an important resource for monitoring the genetic diversity of residual populations, allowing to study several aspects which are still poorly known about the biology of these organisms.
Thanks to our involvement in the National Antarctic Research Program, we extended our comparative genomic studies to diverse polar species, which are extremely interesting due to their adaptation to extreme environments. The use of transcriptomic approaches in the investigation of the molecular mechanisms at the basis of their tolerance to sub-zero temperatures will enable to understand which long-term effects global warming might have on benthonic fish and invertebrates.
Another topic of interest deals with the study of local marine fishes, both of commercial and ecological relevance. We are currently working on the seabass Dicentrachus labrax, trying to elucidate through molecular means its complex immune response, and the abyssal fish Chauliodus sloani, whose physiological adaptation to the deep-sea environment are highly interesting.
Prokaryotic and eukaryotic molecular biodiversity
(Researchers involved: Alberto Pallavicini e Fiorella Florian)
DNA metabarcoding refers to the automated identification of multiple species from a single bulk sample containing entire organisms or from a single environmental sample containing degraded DNA (soil, water, faeces, skin, etc.). Our unit works on the definition of the microbial community of aquacultured bivalves but also, in collaboration with ecologists, on samples of different marine niches for the ecological assessment of microbial and zooplakton communities.
We also work on airborne pollen and fungi spores. For further informations please visit the web page of the research team “Biology of lichen symbiosis”.
In the last years, the team of the Applied and Comparative Genomics laboratory has been involved in the detection and analysis of the environmental DNA (eDNA) in several aquatic compartments, such as freshwater and marine environments. The eDNA consists of traces of DNA released into the environment by the species that live in. With specific molecular methods, the team analyses these traces and is thus able to outline maps of the presence and distribution of the monitored species. To date, eDNA analyses have been applied to the monitoring of the two main species of decapod crayfish, native to the FVG region, for conservation purposes, but also to monitor invasive species of crayfish and fish. As regards the marine habitats, the team is involved in the determination of the composition of the marine zooplankton and in the detection of marine emerging pathogens, such as the Haplosporidium pinnae in the fun mussel Pinna nobilis.
Moreover, these techniques are applied to explore human-microbes interactions. Humans live in constant association with microbes that are present on surfaces and in cavities of the human body, and even within our cells. The number of our microbial companions exceeds by at least ten-fold those of cells of our own body and the number of unique genes they encode is at least 100-fold greater than the number of genes in our own genome. This complex and dynamic microbiota has a profound influence on physiology, nutrition, immunity and development and disruptions in these human-associated microbial communities are a significant factor in many diseases. Defining the dynamic microbial diversity represents the next frontier of genomics. In this contest bacterial metabarcoding analysis are ongoing for gut, vaginal and tooth samples linked to different pathologies.
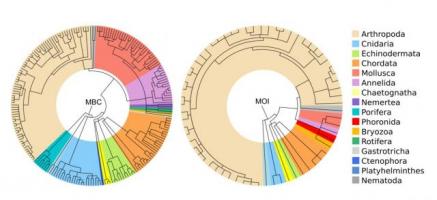
Taxonomic tree representing the marine zooplankton taxonomic richness revealed with metabarcoding (MBC) and morphological inspection (MOI)
Most relevant national and international collaborations in the past 5 years
- Università di Genova (Paola Canesi, Luigi Vezzulli)
- Università Politecnica delle Marche (Adriana Canapa, Antonio Dell’Anno)
- Istituto Nazionale di Oceanografia e Geofisica - OGS (Paola Del Negro, Federica Cerino, Valentina Tirelli)
- Stazione Zoologica Anton Dohrn - SZN (Sergio Stefanni, Immacolata Castellano, Maria Vittoria Modica, Enrico D’Aniello, Maria Sirakov)
- Università della Tuscia (Giuseppe Scapigliati, Francesco Buonocore, Andrea Miccoli)
- Università di Padova (Paola Venier, Marta Giacomello)
- Università di Ferrara (Giorgio Bertorelle)
- National Institute of Biology (David Stankovic)
- University of Split (Tomislav Roncevic)
- CSIC (Antonio Figueras, Beatriz Novoa)
- Paris Natural History Museum (Nicolas Puillandre)
- University of Ljubljana (Gregor Anderluh)
- Yokohama City University (Yasuhiro Ozeki)
- University of Nagasaki (Yuki Fujii)


